
徐华强/谢欣/王明伟/庄友文合作揭示强效镇痛药芬太尼和吗啡的作用机理
日期:2022-11-11 浏览次数:23806
来源:BioArt
疼痛,尤其是慢性疼痛,是一类常见的神经系统疾病。疼痛的影响群体广泛,据统计,全球有近20%的成年人受到慢性疼痛的困扰,在一些经济相对落后的国家更是高达近40%。常见的慢性疼痛包括腰背疼痛、关节炎疼痛、偏头痛以及癌痛等,不仅导致行为能力的减弱或丧失,同时也带来抑郁、睡眠障碍和自杀倾向等不良效应,严重影响人们的身心健康,同时给社会带来了巨大的经济负担。
阿片类药物是目前应用最为广泛且高效的镇痛药物。人类对于阿片类药物的应用可以追溯到几千年之前将植物罂粟用于镇痛镇静和娱乐用途(愉悦、欣快感发生),随后研究发现,阿片类药物吗啡是罂粟里面发挥活性的主要物质。常见的阿片类药物有天然的阿片类生物碱如吗啡、可卡因,以及人工合成的阿片类药物杜冷丁、芬太尼等,合成的阿片类药物在人体内产生吗啡样效应。阿片类药物通过作用于人体内G蛋白偶联受体家族中的阿片受体,尤其是µ型阿片受体(µ opioid receptor, µOR),主要激活下游抑制性Gi/o蛋白发挥镇痛等生理活性。靶向阿片受体药物开发长期以来是镇痛药物研究的热点,已经上市的阿片类药物大多为µOR的激动剂,作为代表性的经典阿片类镇痛药物,吗啡和芬太尼均表现出对µOR的高度选择性。然而,阿片类镇痛药物的使用具有诸多毒副作用,包括成瘾、呼吸抑制、便秘等,极大限制了阿片类镇痛药的临床应用,也使得靶向阿片受体安全有效的镇痛药开发成为了一大难点。阿片类药物成瘾引起的呼吸抑制致死也直接促使了广泛扩散的“阿片危机”的产生,主要集中在北美和加拿大等地,造成每年超10万人死亡,这主要是由于芬太尼及其衍生物滥用导致。作为“阿片危机”的主要产生因素以及目前临床仍在使用的一个强效镇痛药物,芬太尼与其受体µOR相互作用的分子机制长期处于未知状态,解析相关分子机制对于我们合理设计更为安全且高效的芬太尼衍生物类镇痛药意义重大。
前期研究表明,阿片类药物产生的镇痛效应由µOR的G蛋白信号通路负责,而副作用则由arrestin信号通路引起。然而,最近多项研究对该假说提出质疑,认为呼吸抑制等神经毒副作用和arrestin信号无关【1】。尽管质疑存在,前期仍有大量的研究投入到G蛋白偏向性的µOR激动剂药物开发的研究上,以发现高效低毒的靶向µOR镇痛药物【2】。2020年,美国FDA批准了首个,也是目前为止唯一一个基于G蛋白偏向性理念设计出来的靶向µOR镇痛药Oliceridine(TRV130)用于中度到重度疼痛治疗,该药物表现出比吗啡更低的毒副作用。由于对μOR的G蛋白偏好性分子机制认识缺乏,自上述假说提出近20多年以来,μOR的G蛋白偏向性激动剂的发现均通过大规模高通量盲筛获得,这极大阻碍了新型靶向μOR的G蛋白偏向性镇痛药物合理设计和发现。
2022年11月10日,中国科学院上海药物研究所徐华强/庄友文团队、谢欣团队和王明伟团队等合作在Cell上以长文形式在线发表了题为Molecular recognition of morphine and fentanyl by the human μ-opioid receptor 的研究论文。该项研究解析并报道了芬太尼、吗啡以及oliceridine等阿片类镇痛药物分别激活μOR的高分辨率三维结构,首次揭示了芬太尼和吗啡识别并激活μOR的作用机制。该研究进一步结合多种细胞水平功能分析和分子动力学模拟等方法,阐明了芬太尼系列衍生物与药靶μOR的构效关系以及μOR介导arrestin(阻遏素)信号的关键结构决定因素等,系列发现深化了对于μOR信号传导调控的理解和认识,为推动开发新型高效低毒的阿片类阵痛药指明了方向。
本项研究中,研究人员首先通过冷冻电镜技术分别解析了人源μOR结合芬太尼、吗啡和DAMGO等平衡性激动剂(表现出G蛋白和阻遏素双向信号活性)以及TRV130、SR17018、PZM21等G蛋白偏向性激动剂的三维结构,通过分子动力学模拟以及细胞水平功能分析对不同信号活性激动剂活化下μOR的信号传导特性进行表征。研究发现,相比于吗啡,芬太尼在μOR的TM2至TM3近胞外端占据额外的结合口袋,此外,芬太尼的苯胺环侧链与氨基酸残基W295和Y328形成直接的π-π疏水相互作用,这赋予了其比吗啡高达50-100倍的受体激活活性。通过对不同芬太尼衍生物的分子对接和点突变功能验证,研究人员进一步探索了芬太尼及其衍生物与μOR的构效关系,揭示了药物分子与D149、Y150、W135和W320等氨基酸残基不同程度的互作在决定芬太尼及其衍生物(卡芬太尼、苏芬太尼、羟甲芬太尼等)不同活性上的关键作用。对解析的系列结构比对分析以及分子动力学模拟发现,G蛋白偏向性激动剂PZM21等更倾向于结合μOR配体结合口袋的TM2/3一侧,而平衡性激动剂芬太尼等则表现出与μOR跨膜区更为广泛均衡的相互作用,并且使得μOR的胞内端结构域更为紧缩,这有利于μOR与阻遏素的结合,也解释了平衡性激动剂表现阻遏素活性的分子机理。基于这些发现,研究人员还基于芬太尼分子骨架设计了不同活性的新型G蛋白偏向性芬太尼衍生物FBD1和FBD3。
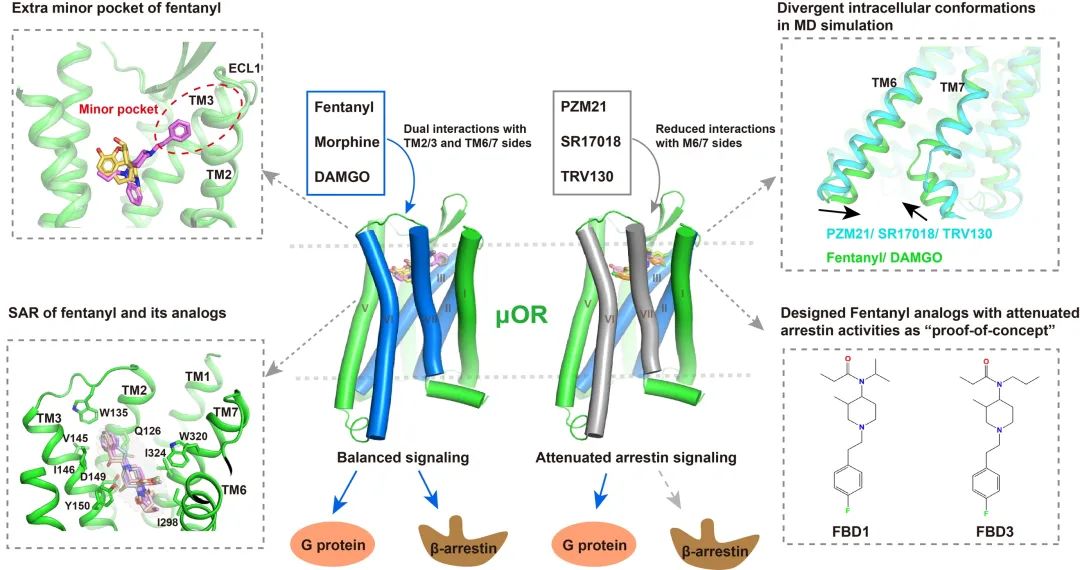
图3. 不同化学结构阿片类药物结合人源μOR的结构。左上:芬太尼和吗啡的不同结合模式;左下:芬太尼及其衍生物与μOR的构效分析;右上:在分子动力学模拟中,μOR的平衡性激动剂相比于G蛋白偏向性激动剂介导更为紧缩的胞内腔构象;右下:基于结构设计的不同活性的新型G蛋白偏向性芬太尼衍生物FBD1和FBD3。中间:减弱配体和μOR的TM6/7的相互作用引起μOR信号偏向性。
本研究由上海药物所徐华强/庄友文团队、谢欣团队和王明伟团队通力合作完成。中国科学院上海药物所副研究员庄友文、博士研究生王悦、何冰清和何欣恒为该论文的共同第一作者。徐华强研究员、谢欣研究员、王明伟教授以及庄友文副研究员为共同通讯作者。
文章链接:https://doi.org/10.1016/j.cell.2022.09.041
 微信公众号
微信公众号
 哔哩哔哩号
哔哩哔哩号
 官方小红书号
官方小红书号
Copyright (c) 2016-2025 中国生物物理学会 版权所有
地址:北京市朝阳区大屯路15号(100101)
电话:会员咨询:010-64888542
学术交流/组织建设:010-64889894
科普宣传:010-64887226
秘书处:010-64887226
传真:010-64889892
E-mail: bscoffice@bsc.org.cn
京ICP备05002793号-2