
姜道华/赵岩报道小分子药物调控人源钠通道NaV1.3的分子机制
日期:2022-03-15 浏览次数:6500
来源:BioArt
在可兴奋细胞中,电压门控钠离子通道 (Voltage-gated sodium channel, NaV) 能够响应膜电位的去极化,引导钠离子快速内流,从而启动和传播动作电位。在哺乳动物中,有9种NaVs亚型 (NaV1.1-1.9) 呈组织特异性分布。其中,NaV1.3主要分布于中枢神经系统中,在胎儿神经元发育中起重要作用。NaV1.3发生突变会导致局灶性癫痫和多小脑回等疾病【1-2】。此外,NaV1.3在受损的外周感觉神经元中重新大量表达,其产生的神经元高兴奋性可能导致神经性疼痛【3】。因此,NaV1.3是抗癫痫和镇痛药物的重要靶点。
几个世纪以来,各种乌头植物 (monkshood) 的根在中国和日本作为传统草药用来止痛、抗风湿以及治疗神经性疾病。草乌甲素 (bulleyaconitine A, 以下简称BLA) 是从滇西乌头 (Aconitum bulleyanum) 中提取的乌头碱 (aconitine) 类活性物质,中国食品药品监督管理局在1985年批准BLA用于治疗类风湿关节炎等引起的慢性疼痛。包括BLA在内的乌头碱(aconitine, from Aconitum napellus)、藜芦定(veratridin, from Liliaceae) 和木藜芦毒素 (grayanotoxin, from Ericaceae) 等植物生物碱,以及箭毒蛙毒素 (batrachotoxin, from frog Phyllobates aurotaenia) 是一组化学结构各异的生物碱,由于这些生物碱对钠通道产生类似的调控效应,因此被归属为site-2神经毒素【4】。site-2神经毒素能够使钠通道的激活电压向去极化方向移动或增加通道开放机率,因此被认为是NaV通道的激动剂;与此同时,乌头碱类毒素使得钠通道的峰值电流显著减小。这类毒素为什么既能“激活”又能“抑制” NaV通道?这一看似“相互矛盾”的调控效应的分子机制仍有待阐明,回答这个问题也有助于生物碱类药物的研发与改良。
目前临床上作用于钠通道的小分子药物通过结合到其中央空腔以阻断钠离子传导,例如局部麻醉剂或抗心律失常药物。然而,由于NaVs的中央空腔区域具有非常高的序列一致性,使得这些药物不可避免的会产生脱靶副作用。因此,寻找新的药物结合位点及筛选具有NaVs亚型选择性药物对减少潜在的脱靶副作用至关重要。ICA-121431 (以下简称ICA) 是一种芳基磺胺类衍生物,能以纳摩尔级别高亲和力选择性地抑制NaV1.3/NaV1.1【5】。然而,NaV1.3如何特异性地识别ICA以及这种拮抗剂对NaV1.3的抑制机制尚不清楚。
2022年3月11日,中国科学院物理研究所姜道华研究员和生物物理研究所赵岩研究员在Nature Communications杂志在线发表了题为Structural basis for modulation of human NaV1.3 by clinical drug and selective antagonist的文章。该研究利用单颗粒冷冻电镜技术解析了人源NaV1.3/β1/β2分别与临床用药BLA结合以及与选择性拮抗剂ICA结合的高分辨率复合物结构,揭示了首个site-2神经毒素的结合位点和一个位于电压感应结构元件(VSDIV)的变构受体位点,阐明了BLA对NaV1.3的激活和抑制机制,以及NaV1.3对ICA的选择性识别机制。
作者首先通过电生理实验表明,BLA更倾向于与开放状态的NaV1.3结合。并且BLA结合一方面使NaV1.3在去极化的电压下更容易激活,另一方面可以抑制NaV1.3的最大电流。NaV1.3/β1/β2与BLA复合物结构清晰地展示了BLA的结合位点——位于中央空腔内靠近domain Ⅰ和domain Ⅱ之间侧窗的位置。通过和开放状态的钠通道结构对比,处于开放状态的结构允许BLA通过其胞内侧激活门孔进入其受体位点。一旦BLA结合到受体位点,它将组成激活门孔的S6Ⅰ和S6Ⅱ稳定在开放构象并阻止其回到静息状态,增加了通道开放的可能性。此外,由于BLA分子相对较大,它部分堵住了中央空腔的离子通道,从而阻挡了钠离子的传导,导致NaV1.3最大电流的显著下降。BLA结合位点的氨基酸在九种钠通道中高度保守,说明此类传统药物缺乏选择性,这可能是脱靶效应导致多种生理副作用的原因。
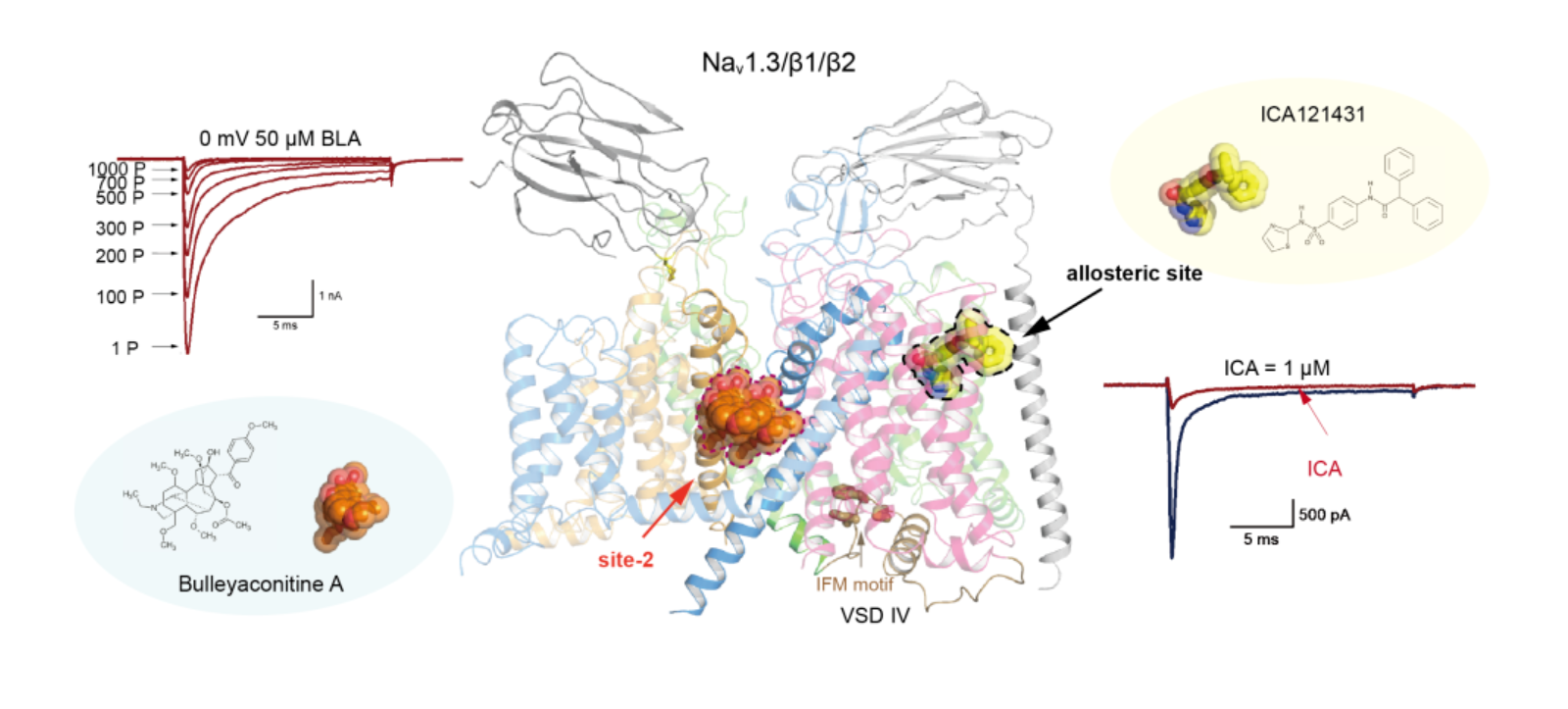
作者通过电生理实验确认了ICA是一种具有钠通道状态依赖抑制特性的抑制剂。为了研究ICA的选择性抑制机制,作者解析了NaV1.3/β1/β2-ICA的冷冻电镜结构。结构分析表明,ICA特异地结合在VSDⅣ的胞外“裂缝”处,将VSDⅣ稳定在激活态构象。通过与NaV1.7选择性抑制剂GX-936复合物结构对比发现,门控电荷R2-R4与这两个药物分子保守的噻唑基团之间的相互作用几乎是一致的,提示R4与芳基磺酰胺负电荷之间的强相互作用是此类抑制剂发挥作用的关键决定因素。结构分析及NaV1.1-1.9的序列比对表明,位于VSDⅣ第二个螺旋上的S1559和R1560是NaV1.3/NaV1.1选择性识别ICA的关键氨基酸。由于ICA将VSDIV稳定在激活状态,导致失活结构元件IFM motif稳定地结合在其受体位点,从而使NaV1.3的激活门孔处于关闭状态。这一机制可能适用于其它抑制NaVs的芳基磺酰胺拮抗剂,因为保守的阴离子头部基团与VSDIV的保守门控电荷结合,决定了它们的抑制作用。与此同时,拮抗剂之间不同的化学结构与NaVs不同的残基相互作用,从而赋予药物亚型选择性。
综上所述,该研究报道了两种化学结构迥异的小分子和NaV1.3不同的结合位点的结构细节,阐明了首个site-2神经毒素BLA对NaV1.3的激活和抑制机制,NaV1.3选择性识别ICA的分子机理及芳基磺酰胺类化合物对NaV的抑制机制,为亚型选择性药物的开发提供了结构基础。
中国科学院物理研究所姜道华研究员和生物物理研究所赵岩研究员为本研究论文的通讯作者。生物物理所研究生李晓静、徐风和徐皓为共同第一作者。中科院生物物理研究所张凯研究员和孙坚原研究员,张树利副研究员为该工作作出重要贡献。中国科学院生物物理研究所生物成像平台为本研究提供了设备和技术支持。
原文链接:https://www.nature.com/articles/s41467-022-28808-5.pdf
参考文献:
5. Mccormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci U S A 2013, 110(29): E2724-E2732.
 微信公众号
微信公众号
 哔哩哔哩号
哔哩哔哩号
 官方小红书号
官方小红书号
Copyright (c) 2016-2025 中国生物物理学会 版权所有
地址:北京市朝阳区大屯路15号(100101)
电话:会员咨询:010-64888542
学术交流/组织建设:010-64889894
科普宣传:010-64887226
秘书处:010-64887226
传真:010-64889892
E-mail: bscoffice@bsc.org.cn
京ICP备05002793号-2